mRNA-Impfstoffe: Warum die Aussagekraft einer Re-Analyse der Zulassungsstudien begrenzt ist
Eine Re-Analyse der Zulassungsstudien von Biontech/Pfizer und Moderna, unter anderem durch den Wissenschaftler Peter Doshi aus den USA, argumentiert, dass ein erhöhtes Risiko für schwere Nebenwirkungen durch Covid-19-Impfstoffe bestehe. Wir haben mit dem Paul-Ehrlich-Institut und einem Statistiker gesprochen. Sie bezweifeln die Aussagekraft der Re-Analyse und weisen auf methodische Mängel hin.

„Schockierende Studien-Ergebnisse, die Sie nicht erfahren sollen!”, titelte der Blog Reitschuster.de am 11. September 2022. Eine erneute Analyse der Zulassungsstudien von Biontech/Pfizer und Moderna belege erhöhte Risiken für schwere Nebenwirkungen durch deren mRNA-Covid-19-Impfstoffe. Als Quelle dient Reitschuster.de ein Interview der Berliner Zeitung (€) mit dem emeritierten Epidemiologen Ulrich Keil. Keil sagte, die erneute Analyse der Zulassungsstudien belege „eine absolute Risikoerhöhung durch die mRNA-Covid-19-Impfung“.
An der Re-Analyse beteiligt war Keil nicht. Sie wurde vom US-amerikanischen Pharmazie-Professor Peter Doshi sowie sechs weiteren Wissenschaftlern durchgeführt und am 22. September in der Fachzeitschrift Vaccine veröffentlicht. Doshi äußerte sich schon 2020 skeptisch über die Zulassungsdaten zu den Impfstoffen. Seine Re-Analyse erregte im September internationales Aufsehen und wurde von der US-amerikanischen Arzneimittelbehörde (FDA) sowie von mehreren Wissenschaftlerinnen und Wissenschaftlern diskutiert.
Wenige Tage nach dem Interview in der Berliner Zeitung führte die Welt ein Interview mit Peter Doshi. Darin erklärt Doshi unter anderem, dass bei den mRNA-Impfstoffen von Biontech/Pfizer und Moderna, beide Studien kombiniert, das Risiko einer schweren Nebenwirkung durch die Covid-19-Impfstoffe um 16 Prozent erhöht sei. Gemeint ist damit, dass bei Geimpften 16 Prozent häufiger Nebenwirkungen auftraten als bei Personen, die nur ein Placebo (Kochsalzlösung) erhielten. In einem Interview mit dem MDR sagte Doshi, die Daten legten nahe, „dass wir bei rund einem von 800 Geimpften ein erhöhtes Risiko schwerer Nebenwirkungen haben“.
Auf Facebook werden Bilder der Medienberichte dutzendfach geteilt; auf Telegram (hier und hier) wird die Re-Analyse als vermeintlicher Beleg interpretiert, dass die Covid-19-Impfung mehr schade als nütze.
Doch die Aussagekraft der Re-Analyse ist umstritten. Im deutschsprachigen Raum kommentierte Impfstoff-Forscher Leif-Erik Sander von der Berliner Charité sie am 22. September auf Twitter mit den Worten: „Die Studie zeigt meiner Meinung nach – nichts.“
Wie also sind die Ergebnisse zu interpretieren? Wir haben das Paul-Ehrlich-Institut und einen Statistiker um ihre Einschätzung gebeten. Sie sagen: Die Re-Analyse weise mehrere methodische Mängel auf; die Ergebnisse seien daher wenig aussagekräftig und belegten nicht, dass Impfungen gegen Covid-19 gefährlicher seien als bisher bekannt.
Re-Analyse bezieht sich auf Daten der ersten Zulassungsstudien von Biontech/Pfizer und Moderna
Vor der Zulassung eines Impfstoffes müssen die Hersteller den Zulassungsbehörden Daten klinischer Studien und Berichte über mögliche Nebenwirkungen zur Verfügung stellen. So auch bei den Impfungen gegen Covid-19. Der Hersteller Biontech/Pfizer hat die Ergebnisse der Phase-3-Studie mit mehr als 43.000 Teilnehmenden im Dezember 2020 im New England Journal of Medicine veröffentlicht. Die Ergebnisse der Phase-3-Studie des Impfstoffes von Moderna mit etwa 30.000 Teilnehmenden wurden im Februar 2021 veröffentlicht. In beiden Studien erhielten jeweils die Hälfte der Teilnehmenden den Impfstoff, die andere Hälfte ein Placebo.
Bei der nun veröffentlichten Re-Analyse wurden die Daten dieser beiden Zulassungsstudien neu interpretiert. Die Wissenschaftler haben also die bereits veröffentlichten Daten erneut analysiert, ohne weitere eigene Daten zu erheben. Die Analyse wendet eine andere Methodik an als die ursprünglichen Zulassungsstudien – und kommt deshalb zu anderen Ergebnissen.
Unterschiedliche Zählweise von potenziellen Impfnebenwirkungen
In den Zulassungsstudien wurde untersucht, ob es durch die Impfstoffe zu sogenannten schweren unerwünschten Ereignissen kommt (serious adverse events, SAE). Diese Ereignisse müssen keine Folge der Covid-19-Impfung sein, die Dokumentation aller Ereignisse bei Teilnehmenden der Studien dient aber dazu, Nebenwirkungen zu erkennen. Wenn ein bestimmtes Ereignis bei den geimpften Probanden der Studie häufiger vorkommt als bei denen, die nur ein Placebo erhielten, ist das ein Hinweis, dass es sich um eine Nebenwirkung der Impfung handelt.
Was sind Serious Adverse Events (SAE)?
Unerwünschte Ereignisse wurden auch in der Re-Analyse untersucht, jedoch mit folgendem Unterschied: Die US-amerikanische Arzneimittelbehörde FDA zählte in den Studien von Biontech/Pfizer und Moderna jeweils die Anzahl der Personen, bei denen ein oder mehrere SAEs aufgetreten sind. In der Re-Analyse wurde hingegen die Gesamtzahl der SAEs gezählt. Wenn also bei einer Person zwei Nebenwirkungen auftraten, wurden diese von der FDA einmal gezählt; in der Re-Analyse hingegen doppelt.
Wie wirkt sich die andere Zählweise aus?
Dadurch kommt die Re-Analyse auf höhere Werte. So ergaben sich bei der Re-Analyse beispielsweise 127 schwere unerwünschte Ereignisse pro 10.000 Probanden mit dem Biontech/Pfizer-Impfstoff; in der Zulassungsstudie hingegen nur 126 schwere unerwünschte Ereignisse unter allen 21.621 Geimpften.
Die Autoren der Zulassungsstudie von Biontech/Pfizer schreiben daher: „Die Häufigkeit schwerer unerwünschter Ereignisse war in der Impfstoff- und der Placebogruppe ähnlich (0,6 Prozent bzw. 0,5 Prozent).“ Da in der Re-Analyse die unerwünschten Ereignisse aber anders gezählt wurden, errechnete diese, dass das Risiko der Geimpften um 36 Prozent höher sei als das der Placebo-Gruppe.
Für den Moderna-Impfstoff gilt dasselbe: Unter 15.185 Geimpften gab es laut der Zulassungsstudie 93 Personen (0,6 Prozent), bei denen ein unerwünschtes Ereignis aufgetreten ist. Unter 15.116 Personen, die das Placebo erhalten hatten, trat bei 89 Personen ein unerwünschtes Ereignis auf (0,6 Prozent). Mit ihrer anderen Zählweise errechnete die Re-Analyse aber, dass die Geimpften ein 6 Prozent höheres Risiko hätten.
Die in der Re-Analyse genutzte Zählweise bewertete zum Beispiel Adam Jacobs, leitender Direktor für Biostatistik bei Premier Research, als eher unüblich. Jacobs sagte gegenüber der britischen Faktencheck-Organisation Full Fact: „Bei der Analyse von unerwünschten Ereignissen ist es weitaus üblicher, die Anzahl der Patienten mit Ereignissen zu analysieren.“ Es sei nicht immer möglich zu wissen, ob es sich um getrennte Ereignisse handele, oder um einen einzigen pathologischen Prozess mit wiederholten Krankheitsschüben.
Ein Argument der Re-Analyse, aus der sich die These von „mehr Schaden als Nutzen“ ableitet, ist, dass das Risiko für eine schwere Nebenwirkung nach der Impfung angeblich höher sei als das Risiko, wegen Covid-19 hospitalisiert zu werden. Die andere Zählweise der unerwünschten Ereignisse könnte bei diesem Vergleich eine Verzerrung bewirken, wie Full Fact an einem Beispiel erklärt: Eine Person, die nach einer Covid-19-Impfung sowohl über Durchfall als auch Bauchschmerzen klage, würde laut der Methode der Re-Analyse zweimal gezählt, während eine Person, die hospitalisiert wird, immer nur einmal zähle.
Peter Doshi räumte gegenüber Full Fact ein, dass „beide Ansätze ihre Berechtigung haben“. Es sei aber nicht möglich gewesen, die Analyse der Probanden mit einer SAE abzuschließen, da er keinen Zugang zu den vollständigen Daten der Zulassungsstudien gehabt habe.
Lediglich öffentlich verfügbare Daten in Re-Analyse einbezogen
Wie uns das Paul-Ehrlich-Institut (PEI) auf Anfrage mitteilte, sei eine Einschränkung der Re-Analyse, dass nur öffentlich zugängliche Daten analysiert werden konnten. Das heißt, individuelle Vorgeschichten oder Erkrankungen der Teilnehmenden sowie deren Alter standen den Autoren nicht zur Verfügung.
Solche Daten seien aber wichtig, weil sich sonst nicht genau sagen lasse, ob unerwünschte Ereignisse von besonderem Interesse (Adverse Events of Special Interest, AESI) in einen plausiblen Zusammenhang mit der Impfung gebracht werden könnten oder nicht. Als Grundlage für eine wissenschaftliche Aussage sei dies zumindest „fragwürdig“, so das PEI.
Zum Beispiel haben Menschen, die älter sind oder bestimmte Vorerkrankungen haben, ein generell erhöhtes Risiko für Herz- oder Kreislauferkrankungen. Ist das nicht bekannt, kann es zu falschen Zuordnungen von zufälligen Ereignissen als Nebenwirkung der Impfung kommen.
Richtig ist, wie die Autoren der Re-Analyse selbst erklären, dass die vollständigen Daten der Zulassungsstudien von Biontech/Pfizer und Moderna nicht öffentlich einsehbar sind. Immer wieder wurde eine Veröffentlichung der Daten gefordert. Doshi gehört zu einer Gruppe, die im August 2021 von der FDA die Freigabe aller Daten und Informationen zu dem Impfstoff von Pfizer/Biontech forderte.
Warum Fachleute Mängel in der Methodik der Re-Analyse sehen: Breite Konfidenzintervalle
Die Re-Analyse kam zu dem Ergebnis: Schwere unerwünschte Ereignisse seien in der Gruppe der Probanden mit dem Impfstoff von Biontech/Pfizer um 36 Prozent häufiger aufgetreten als in der Placebogruppe. Bei Moderna seien es 6 Prozent gewesen. Beide Studien kombiniert zeigten ein um 16 Prozent erhöhtes Risiko einer schweren Nebenwirkung durch die mRNA-Impfstoffe.
Wir haben Christoph Rothe, Inhaber des Lehrstuhls für Statistik an der Universität Mannheim, um eine Einschätzung der Daten gebeten. Er sagte uns, ein Problem der Re-Analyse sei, dass sie sehr breite Konfidenzintervalle aufweise.
Ein Konfidenzintervall (confidence interval, CI) gibt den Bereich an, in dem sich zum Beispiel in 95 Prozent der Fälle der sogenannte wahre Wert befindet.
Ein Beispiel: Anhand der Daten, wie viel eine Stichprobe von 100 Menschen für Pizza ausgeben, könnte man versuchen, die durchschnittlichen Ausgaben aller Menschen in Deutschland für Pizza zu schätzen. Berechnet man das 95%-Konfidenzintervall anhand mathematischer Formeln, erhält man eine Spannbreite (Ober- und Untergrenze der wahren Pizza-Ausgaben). Das heißt: In 95 Prozent der Fälle liegen die durchschnittlichen Ausgaben für Pizza aller Menschen in Deutschland zwischen diesen beiden Werten.
Übertragen auf die Berechnung des Risikos für Impfnebenwirkung heißt das, dass der wahre Wert mit einer Wahrscheinlichkeit von 95 Prozent in dem errechneten Bereich liegt.
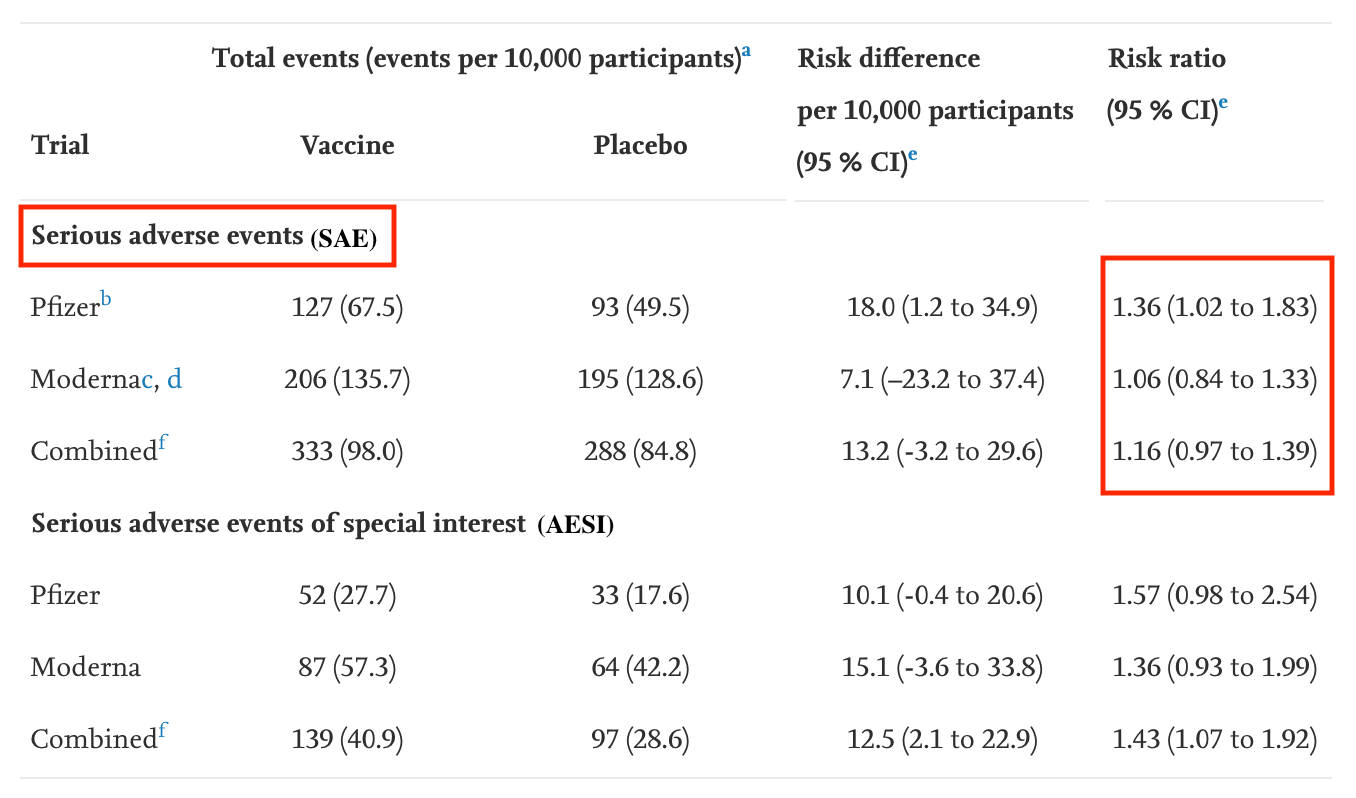
In der Re-Analyse wurde zum Beispiel das Risiko schwerer Impfnebenwirkungen in beiden Studien kombiniert mit einem sehr breiten Bereich zwischen 0.97 und 1.39 angegeben. 0.97 bedeutet, dass das Risiko der Geimpften drei Prozent niedriger wäre als das der Placebogruppe – 1.39 bedeutet, es wäre 39 Prozent höher. In dieser Spannbreite liegt der wahre Wert.
Der Statistiker Christoph Rothe erklärte uns: „Das bedeutet grob gesagt, dass mit Blick auf die Daten alles zwischen einer Reduktion um drei Prozent und einem Anstieg um 39 Prozent als wahrer Wert für die Risikoänderung nicht unplausibel erscheint.“ Anders gesagt: Die Schätzung ist unpräzise.
Ein um 16 Prozent erhöhtes Risiko für schwere Nebenwirkungen bei Geimpften könnte ein Zufallsergebnis sein. Eine Unsicherheit, die dazu führt, dass die Daten der Re-Analyse nicht belegen können, ob eine Covid-19-Impfung „mehr schadet als nützt“.
Re-Analyse untersucht andere „unerwünschte Ereignisse“ als die Studien von Biontech/Pfizer und Moderna
Im zweiten Schritt haben die Wissenschaftler der Re-Analyse eine Auswahl von schweren unerwünschten Ereignissen getroffen: „schwere unerwünschte Ereignisse von besonderem Interesse“ (AESI). Dafür haben sie alle SAEs mit einer externen Liste, die speziell für Covid-19-Impfungen erstellt wurde, abgeglichen. Dadurch wurden viele Ereignisse ausgeschlossen. Welche das sind, zeigt eine Liste mit Beispielen sowie eine Tabelle im Anhang der Re-Analyse (Download).
Auf dieser Grundlage berechneten die Autoren einen weiteren Risikowert, das Risiko für ein AESI. Es liegt der Re-Analyse zufolge für Geimpfte mit dem Biontech/Pfizer-Impfstoff um 57 Prozent und für Geimpfte mit dem Moderna-Impfstoff um 36 Prozent höher als für Ungeimpfte. Als AESI zählten die Autoren zum Beispiel unerwünschte Ereignisse wie Bauchschmerzen, Oberbauchschmerzen, akutes Atemversagen oder Hautausschlag.
Woher stammt die Liste mit schweren unerwünschten Ereignissen von besonderem Interesse (AESI)?
Dieses Vorgehen kritisieren das Paul-Ehrlich-Institut und die US-amerikanische Arzneimittelbehörde FDA. In der Kritik der FDA (Download) heißt es, die Interpretation von schweren Nebenwirkungen könne nicht zuverlässig anhand der Brighton Collaboration Liste erfolgen. David Gorski, Professor für Chirurgie und Onkologie an der Wayne State University School of Medicine, schrieb in einem Blogbeitrag am 5. September, manche unerwünschten Ereignisse auf der Ausschlussliste erschienen sinnvoll, wie zum Beispiel Schusswunden oder Kopfverletzungen. Der Ausschluss anderer Ereignisse sei aber fragwürdig, wie zum Beispiel Erbrechen oder Darmverdrehung.
Das Paul-Ehrlich-Institut schrieb uns: „Beim Erstellen der AESI-Liste [war] der Fokus auf die Komplikationen und Symptome der Covid-19-Erkrankung gerichtet. Wenn man in der Darstellung der Methoden angibt, Impfrisiken betrachten zu wollen, führt dieser Blickwinkel zu weiteren Verzerrungen.“
Unpräzise Messungen verringern Aussagekraft der Re-Analyse
Die Konfidenzintervalle der AESIs sind, wie von Christoph Rothe beschrieben, wieder sehr breit. Dadurch sind auch hier die Ergebnisse der statistischen Schätzung mit Unsicherheiten behaftet.
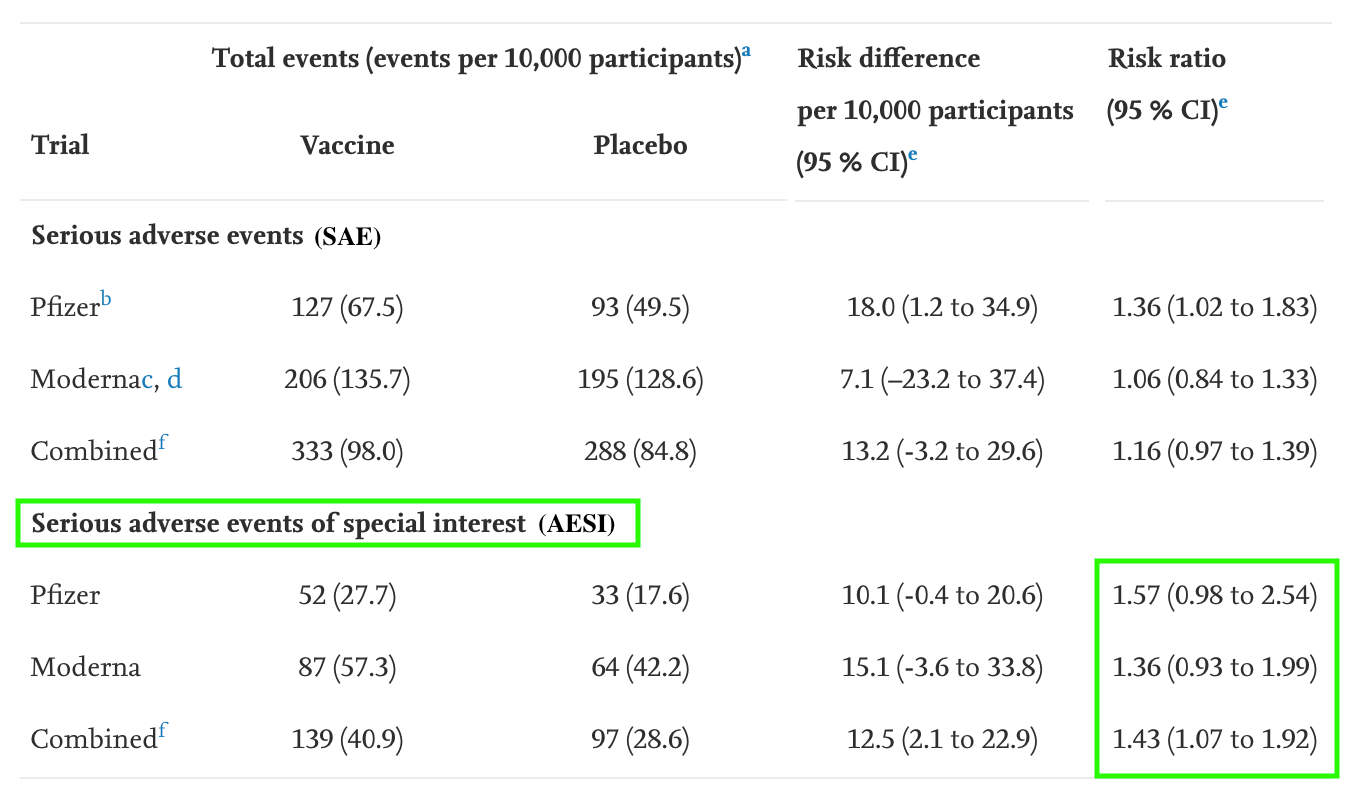
Laut Rothe erlaubten die Daten, die in der Re-Analyse untersucht wurden, „die präzise Messung eines Effektes nicht“. Durch die Definition, was genau als AESI zählt, hätten die Autoren der Re-Analyse zudem einen gewissen Spielraum, um das Ergebnis der Analyse zu beeinflussen.
Autoren der Re-Analyse kritisieren Entblindung der Zulassungsstudien – dies war jedoch der Wunsch vieler Teilnehmender
Die Autoren der Re-Analyse kritisieren außerdem, dass nach der Notfallzulassung der beiden mRNA-Impfstoffe durch die US-Arzneimittelbehörde (FDA) die Zulassungsstudien von Biontech/Pfizer und Moderna „entblindet“ wurden. Das heißt, den Teilnehmenden wurde nachträglich gesagt, ob sie einen Impfstoff erhalten hatten oder nur ein Placebo.
In einem offenen Brief forderten tatsächlich mehr als 300 Teilnehmende der Zulassungsstudien die vorläufige Entblindung aller Personen aus der Placebo-Gruppe, die laut den Impfplänen der Regierung an der Reihe wären, sich impfen zu lassen. Sie bezogen sich dabei unter anderem auf den Ethikkodex der American Medical Association.
Wir haben bei der Geschäftsstelle des deutschen Ethikrats nachgefragt, ob es ungewöhnlich gewesen sei, die Studie zu diesem Zeitpunkt zu entblinden. Die Geschäftsstelle schrieb uns, dass dies allgemein kein ungewöhnlicher Vorgang sei: „Aus ethischen Gründen ist eine Entblindung insbesondere dann sinnvoll oder sogar geboten, wenn sich im Studienverlauf entweder Hinweise auf gravierende Nebenwirkungen des Verum-Präparats [Impfstoff, Anm. d. Red.] ergeben oder sich dieses im Gegenteil als so nutzbringend darstellt, dass es nicht rechtfertigbar erscheint, es den Probanden im Placebo-Arm der Studie länger vorzuenthalten.”
Die zweite genannte Möglichkeit gewinne umso mehr an Gewicht, je dringender die medizinische Notlage sei, für die ein Präparat oder eine Impfung Abhilfe verspreche. Zum Zeitpunkt der Entblindung, Anfang 2021, wurden weltweit über 84 Millionen Covid-19-Erkrankungen und etwa 1.9 Millionen Covid-19-Todesfälle der Weltgesundheitsorganisation (WHO) gemeldet (Report, Stand: 31. Dezember 2020).
Wie in einem Situationsbericht der WHO (PDF) vom 18. Dezember 2020 zu lesen ist, hatten die FDA und die europäische Arzneimittelagentur (EMA) bereits signalisiert: Bei entsprechenden vorläufigen Ergebnissen, und wenn Teilnehmende eine Entblindung vor dem Ende der Phase-3-Zulassungstudien wünschen, sei es „die ethische Pflicht, dieser Bitte nachzukommen“.
Re-Analyse bezieht sich auf Daten von 2020 – seitdem wurden Nebenwirkungen der Impfstoffe in zahlreichen anderen Studien untersucht
Das Paul-Ehrlich-Institut teilte uns mit: Aus den weitreichenden Erfahrungen bei der Anwendung der Covid-19-Impfstoffe, ergebe sich auch vor dem Hintergrund der Re-Analyse „kein Anlass für eine Änderung der Bewertung des günstigen Nutzen/Risiko-Verhältnisses dieser Impfstoffe“.
Die Daten der Re-Analyse beziehen sich auf den Stand Ende 2020 beziehungsweise Anfang 2021. Seitdem wurden viele weitere Studien (hier, hier und hier) durchgeführt, die die Sicherheit und potentielle Nebenwirkungen der Impfstoffe untersucht haben. Zum Beispiel untersuchte eine Studie aus Israel, die am 16. September 2021 im New England Journal of Medicine veröffentlicht wurde, die sogenannte „Real World Evidence“: Daten von insgesamt knapp 1,8 Millionen Menschen in der alltäglichen Praxis. Die Ergebnisse zeigen, dass der mRNA-Impfstoff von Biontech/Pfizer in keiner Verbindung mit den meisten der unerwünschten Ereignisse steht; etwa drei von 100.000 Geimpften haben eine Herzmuskelentzündung erlitten.
Der Statistiker Christoph Rothe schrieb uns: „Von einer Re-Analyse der in dieser Hinsicht überschaubaren Daten aus den Zulassungsstudien, war daher von vornherein kein großer Erkenntnisgewinn zu erwarten.“
Fazit: In Medienberichten, Blogartikeln und Sozialen Netzwerken wurde der Re-Analyse der Zulassungsstudien für die mRNA-Impfstoffe gegen Covid-19 mehr Aussagekraft zugesprochen, als letztlich dahinter steckt. Fachleute kritisieren die Auswahl und Zählweise der untersuchten mutmaßlichen Impfnebenwirkungen. Zudem sind die Risikounterschiede mit Unsicherheiten behaftet, das heißt, die Ergebnisse könnten Zufall sein.
Die Autoren der Re-Analyse kritisieren die frühzeitige Entblindung der Zulassungsstudien von Biontech/Pfizer und Moderna. Sie erwähnen jedoch nicht, dass die Teilnehmenden selbst in einem offenen Brief eine Entblindung forderten und es als ethische Pflicht angesehen werden kann, dieser Forderung nachzukommen.
Update, 16. November 2022: Wir haben die Definition eines Konfidenzintervalls präzisiert.
Update, 15. November 2022: Wir haben in der Einleitung präzisiert, mit welchen Quellen wir für diesen Faktencheck gesprochen haben.
Mitarbeit: Sophie Timmermann
Redigatur: Matthias Bau, Alice Echtermann
Die wichtigsten, öffentlichen Quellen für diesen Faktencheck:
- Re-Analyse u.a. von Peter Doshi, „Serious adverse events of special interest following mRNA COVID-19 vaccination in randomized trials in adults“, Vaccine, 22. September 2022: Link (archiviert)
- Weitere Informationen zu Fairman et al.: Antwort auf die Kritik der FDA: Link (Download)
- Blogartikel von David Gorski „Peer review fail: Vaccine published antivax propaganda disguised as „reanalyses“ of Pfizer and Moderna Covid-19 vaccine clinical trial data“, 5. September 2022: Link
- Emergency Use Designation of Covid-19 candidate vaccines: Ethical considerations for current and future Covid-19 placebo-controlled vaccine trials and trial unblinding, WHO: Link
- Offener Brief der Teilnehmenden der Zulassungsstudien, November 2020: Link
- Zulassungsstudie Biontech-Impfstoff, „Safety of the BNT162b2 mRNA Covid-19 Vaccine in a Nationwide Setting“, New England Journal of Medicine, 16. September 2021: Link
- Zulassungsstudie Moderna-Impfstoff „Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine“, New England Journal of Medicine, 4. Februar 2021: Link


